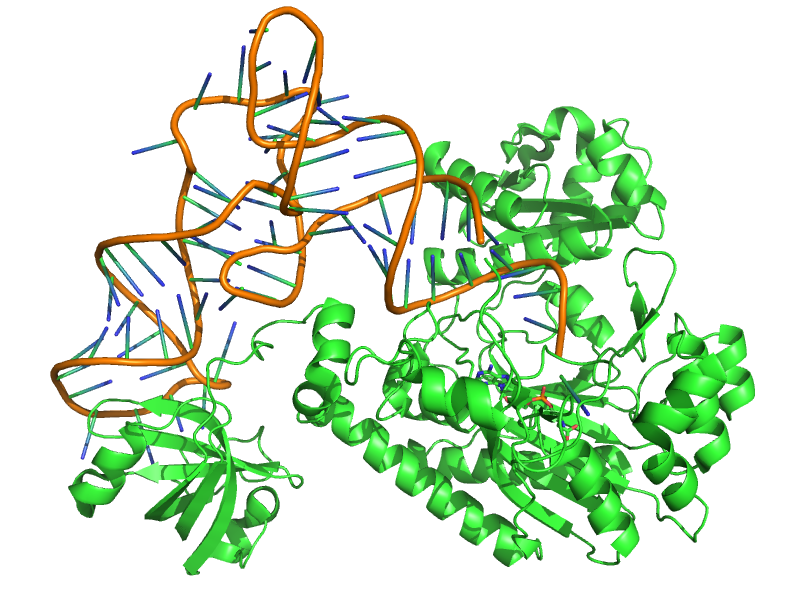
L'objectif du TP est d'étudier par modélisation moléculaire la reconnaissance spécifique entre l'aspartyl-ARNt synthétase et son substrat Asp. On cherchera à évaluer la spécificité en comparant la fixation des ligands Asp et Asn. On essaiera ensuite d'identifier et de modéliser des mutations dans le site actif qui pourraient favoriser la fixation d'Asn à la place d'Asp. C'est un premier pas vers une ingénierie du code génétique.
Les aminoacyl-ARNt synthétases (aaRS) constituent une famille d'enzymes impliquées dans la synthèse protéique. Elles interviennent au niveau de la traduction en permettant la liaison de l'acide aminé à son ARN de transfert. Elles sont très spécifiques de l'acide aminé concerné et de l'ARN de transfert qui lui correspond. Il en existe donc une pour chaque acide aminé.
Nous allons nous intéresser plus particulièrement à l'aspartyl-ARNt synthétase (AspRS), le but étant d'effectuer des mutations ponctuelles sur cette enzyme afin de réduire son affinité pour son ligand naturel aspartate et favoriser sa liaison avec l'asparagine.
Pour cela, nous envisagerons le problème en terme de séquences et de structures protéiques. L'étude comprend trois étapes :
Cette analyse conduira à proposer des mutations judicieuses du site actif, permettant de modifier la spécificité de l'AspRS en privilégiant la liaison de l'Asn à la place de l'Asp.
L'AspRS d'E. coli comporte trois domaines : celui de la fixation de l'anticodon de l'ARNt, celui du site catalytique, et un troisième domaine inséré dans celui du site catalytique.
Lancer une recherche BLAST. Quels types de protéines trouve-t-on ?
Identifier des régions fortement conservées qui peuvent correspondre au site actif. Choisir quelques positions qui semblent caractéristiques de l'AspRS et de la fixation de l'Asp.
Quelle stratégie avez-vous employée ? Quelles mutations proposez-vous pour modifier l'affinité de l'AspRS pour l'aspartate et l'asparagine ?
Avec les informations obtenues précédemment, proposer des mutations judicieuses pour modifier l'affinité de l'AspRS pour l'aspartate et l'asparagine. On s'efforcera d'en tester plusieurs dans l'étape ultérieure de modélisation.
L'inspection du site actif vous conduit-elle à modifier vos propositions de mutations faites à partir des séquences ?
Peut-on utiliser la structure pour vérifier l'alignement des séquences ?
C'est la partie la plus ambitieuse et complexe du TP. Il y a deux étapes :
On suivra le protocole suivant avec le programme XPLOR :
Les fichiers PDB doivent respecter un format particulier pour être lisibles par XPLOR. Le nom du segment doit être placé sur 4 caractères dans les colonnes 73-76. On remarque également que le code à 3 lettres des histidines a été changé de HIS en HIE. Il existe en effet 3 états de protonation possibles pour les histidines et il est nécessaire de préciser à XPLOR quel état est choisi parmi HID, HIE ou HIP (voir le fichier de topologie amber.rtf pour la définition de ces états).
L'état de protonation HIE choisi pour toutes les histidines vous semble-t-il raisonnable ? En cas de doute, tester d'autres états de protonation et évaluer l'impact sur les résultats.
xplor < build.inp > build.out
xplor < minimize.inp > minimize.out
xplor < energy.inp > energy.out
Quelle est l'affinité de la protéine AspRS pour le ligand Asp ?
Quelle est l'affinité de l'AspRS pour Asn ?
Expérimentalement, l'enzyme sauvage fixe Asp nettement mieux que Asn, avec une différence d'énergie libre d'association de plus de 7 kcal/mol. Retrouvez-vous cette tendance ?
Une mutation simple (par exemple Asp→Asn ou Gln→Glu) peut être réalisée par édition du fichier PDB, comme expliqué pour le ligand.
Une mutation plus complexe pourra être effectuée avec le programme SCWRL. Le choix de la mutation (par exemple R10K) se fera en remplaçant dans le fichier asprs.seq le code à une lettre de l'acide aminé natif en minuscule par celui de l'acide aminé choisi pour la mutation en majuscule (par exemple remplacer le « r » minuscule en position 10 par un « K » majuscule).
On lance ensuite le programme SCWRL de la manière suivante :
scwrl -s asprs.seq -i asprs.wt.pdb -o asprs.pdb > scwrl.out
Comparer la structure mutée obtenue asprs.pdb avec la structure native asprs.wt.pdb.
Il est conseillé de travailler dans un dossier séparé pour chaque mutant.
Pour utiliser la structure mutée par SCWRL avec XPLOR, il faudra s'assurer qu'elle soit correctement formatée. On utilisera pour cela le programme pdb2xplor de la façon suivante :
pdb2xplor asprs.pdb A PROT > asprs.xplor.pdb
Quelles affinités pour Asp et Asn obtenez-vous avec la protéine mutée ?
Avez-vous réussi à inverser la spécificité ?
Interpréter structuralement l'effet des mutations.
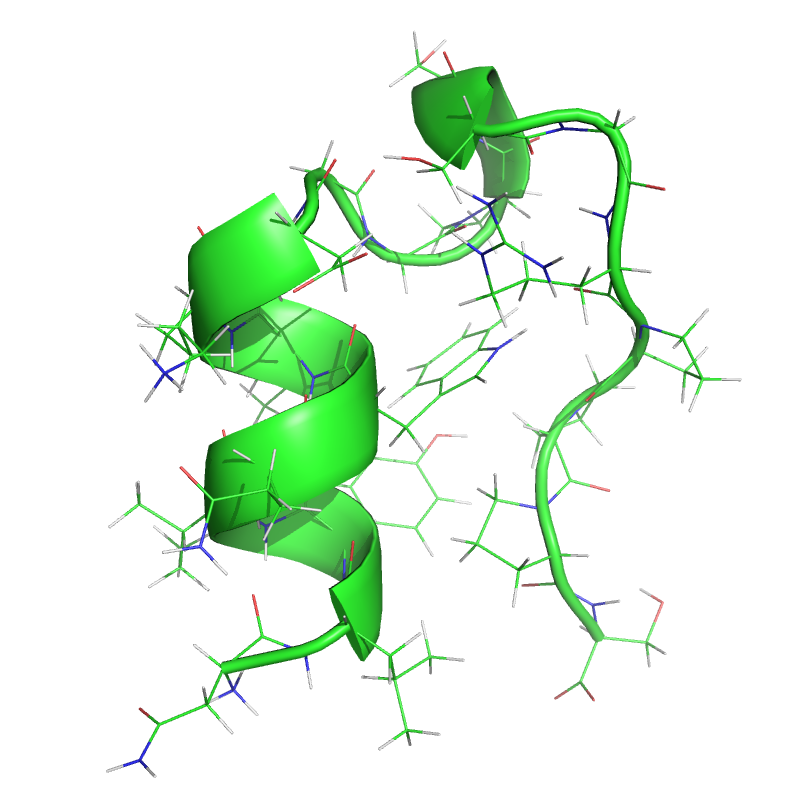
L'objectif du TP est d'étudier la structure et la stabilité d'une petite protéine, le Trp-cage.
 ↔
↔
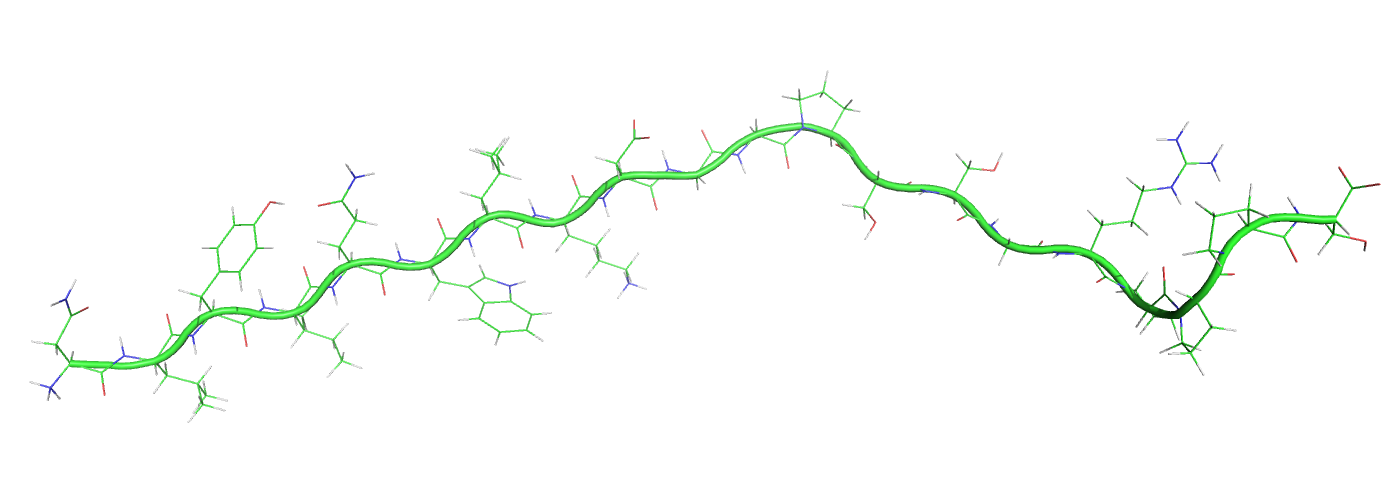
Le Trp-cage est une petite protéine artificielle de 20 acides aminés, qui a été conçue pour se replier facilement. Sa séquence d'acides aminés est NLYIQWLKDGGPSSGRPPPS. Le problème du repliement des protéines compte parmi les défis les plus importants de la bioinformatique structurale. Il consiste à prédire la structure tridimensionnelle d'une protéine à partir uniquement de l'information de sa séquence.
Nous emploierons les méthodes de la mécanique moléculaire pour modéliser le Trp-cage.
xplor < build.inp > build.out
Ce script construit un modèle du Trp-cage avec XPLOR et effectue une minimisation de l'énergie pour améliorer la géométrie.
Examiner le fichier de sortie et visualiser les structures produites.
xplor < md.inp > md.out
Ce script effectue une dynamique moléculaire du Trp-cage pendant 20ps en assignant des vitesses initiales aléatoires puis en maintenant la température à 300K.
Examiner le fichier de sortie et suivre l'énergie et la température en fonction du temps.
xplor < traj2mpdb.inp > traj2mpdb.out
Ce script convertit le format de la trajectoire produite de DCD à PDB multiple.
On pourra ensuite visualiser la trajectoire avec PyMOL en la chargeant de la façon suivante :
load md.multi.pdb, multiplex=0
xplor < analyze.inp > analyze.out
Ce script lit la trajectoire produite (md.dcd) et effectue des calculs structuraux ou énergétiques à chaque pas. Les résultats sont écrits dans un fichier texte (md.dat). Les représenter graphiquement.
Les analyses incluses dans le script ne le sont qu'à titre d'exemple, à vous d'en ajouter d'autres plus pertinentes en vous aidant de la documentation d'XPLOR.
Le mimivirus est un virus à ADN géant. Il est de taille supérieure à de nombreuses bactéries et peut lui-même être infecté par d'autres virus. On a découvert que le mimivirus possédait certains gènes de protéines impliquées dans la traduction, absents dans les autres virus qui utilisent la machinerie de la cellule hôte pour se multiplier. Ces découvertes ont alimenté les débats sur la frontière entre matière vivante et inerte.
La modélisation par homologie a pour objet de construire un modèle de la structure inconnue d'une protéine cible (« target »), connaissant sa séquence et la structure d'une autre protéine guide (« template ») de séquence homologue. La méthode peut se décomposer en quatre étapes :
Le but de ce travail est de proposer le meilleur modèle structural possible (critère à définir) de la tyrosyl-ARNt synthétase de mimivirus (on suppose sa structure inconnue) par modélisation par homologie avec le programme Modeller.
Récupérer la séquence de la tyrosyl-ARNt synthétase de mimivirus au format FASTA dans la base de données UniProt (http://www.uniprot.org).
Sélectionner judicieusement une structure qui servira de guide pour la modélisation par homologie (on s'abstiendra bien-entendu de prendre la structure de la tyrosyl-ARNt synthétase de mimivirus que l'on suppose inconnue). Récupérer cette structure au format PDB.
Convertir la séquence requête du format FASTA vers le format PIR (http://salilab.org/modeller/manual, File formats, Alignment file (PIR)) avec lequel Modeller travaille. Exemple d'une séquence au format PIR :
>P1;TvLDH sequence:TvLDH:::::::: MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLAGFVATTDPKA AFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGNPDNTNCEIAMLHAKNLKPEN FSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLTQATFTKEGKTQKVVDVLDHDYVFDTFFKKI GHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTAPGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVV EGFKVNDWLREKLDFTEKDLFHEKEIALNHLAQGG*
Le programme Modeller se lance de la façon suivante :
modeller file.py
Aligner la séquence requête avec la séquence de la structure guide sélectionnée en adaptant le script align2d.py.
modeller align2d.py
L'alignement produit est écrit aux formats PIR, PAP et FASTA. Examiner ces fichiers.
Modéliser par homologie la structure cible en adaptant le script model-single.py.
modeller model-single.py
Un résumé comprenant le nom des fichiers PDB des modèles produits ainsi que la valeur de la fonction d'énergie de Modeller et le score DOPE pour chaque modèle se trouve à la fin du fichier de sortie (model-single.log). Examiner les modèles produits avec PyMOL.
Évaluer les modèles générés à l'étape précédente en adaptant le script evaluate_model.py.
modeller evaluate_model.py
Ce script permet une évaluation plus détaillée des modèles produits en calculant le score DOPE pour chaque position de l'alignement. Tracer le score DOPE en fonction de la position (colonnes 1 et 42) pour les différents modèles.
Pour aller plus loin :
Modeller tutorial : http://salilab.org/modeller/tutorial/basic.html
Modeller manual : http://salilab.org/modeller/manual
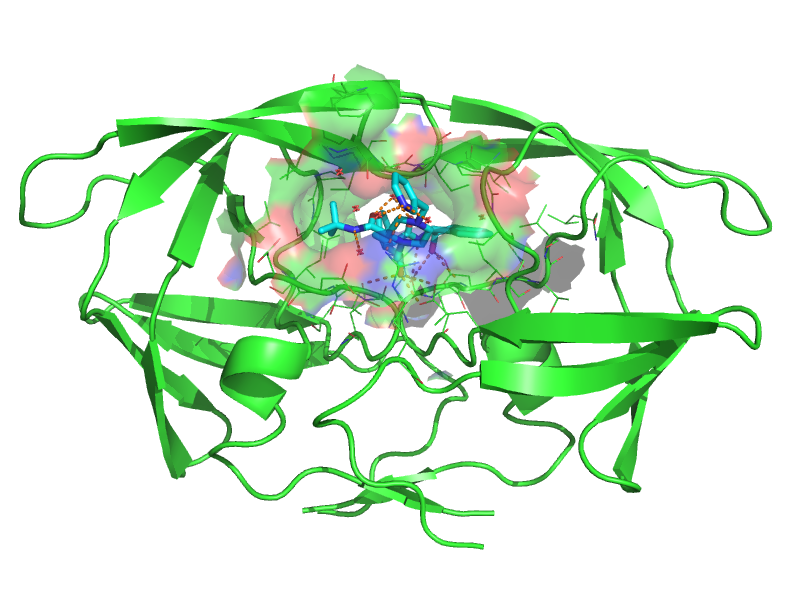
AutoDock est une suite d'outils destinés à l'amarrage moléculaire (« molecular docking »). Le docking consiste à prédire comment des ligands, comme des substrats ou des médicaments potentiels, se fixent sur un récepteur de structure tridimensionnelle connue.
La procédure de docking avec AutoDock se décompose en deux étapes principales :
Nous utiliserons également une interface graphique appelée AutoDockTools (ADT) pour faciliter la préparation du docking et la visualisation des résultats.
La méthode sera illustrée avec le docking de l'Indinavir, un inhibiteur de la protéase du VIH-1, utilisé comme antirétroviral dans le traitement du SIDA.
AutoDock a besoin de connaître les charges et types atomiques de chaque atome, ainsi qu'une liste des liaisons avec libre rotation présentes dans le ligand.
adt
→ → → → "ind.pdb"
AutoDockTools lit le ligand et effectue les étapes suivantes : calcul des charges atomiques de type Gasteiger, fusion des hydrogènes non-polaires, attribution des types atomiques, détection du nombre de degrés de liberté en torsion.
Les types atomiques et les charges sont utilisés dans les termes de mécanique moléculaire de la fonction de score d'AutoDock. Le nombre de degrés de liberté en torsion du ligand détermine sa flexibilité et intervient également dans le calcul de la pénalité entropique d'association.
→ →
Le plus petit groupe rigide de la molécule inclut cet atome et tous les atomes connectés à lui par des liaisons sans libre rotation.
→ →
Les liaisons sans libre rotation apparaissent en rouge, celles qui pourraient subir une rotation mais qui sont marquées comme inactives apparaissent en violet, enfin les liaisons marquées comme actives apparaissent en vert. Laisser la définition par défaut qui correspond à 14 degrés de liberté.
→ →
Réduire le nombre de degrés de liberté à 6 ("fewest atoms") pour accélérer le calcul.
→ → → "ind.pdbqt"
→ → → → "hsg1.pdb"
AutoDockTools lit le récepteur et comme pour le ligand effectue les étapes de calcul des charges atomiques de type Gasteiger, fusion des hydrogènes non-polaires et attribution des types atomiques.
Sauvegarder le récepteur sous le nom "hsg1.pdbqt".
Il est nécessaire de générer une carte d'interaction pour chaque type atomique du ligand plus une carte pour l'électrostatique et une carte pour la désolvatation.
→ → → "ind" →
→
Choisir 60, 60 et 66 pour le nombre de points de la grille dans les directions x, y et z ; et 2.5, 6.5 et -7.5 pour les coordonnées x, y et z de la position du centre de la grille.
→
→ → → "hsg1.gpf"
Le fichier .gpf contient les paramètres pour le programme AutoGrid.
→ →
Le programme AutoGrid est exécuté avec le fichier de paramètres
généré précédemment (la ligne de commande correspondante est
autogrid4 -p hsg1.gpf -l hsg1.glg).
Les cartes produites sont écrites dans le répertoire courant et ont l'extension .map.
→ → → "hsg1.pdbqt"
→ → → "ind" → →
→ →
Réduire le nombre d'évaluations de l'algorithme génétique à 250000 ("short") puis cliquer sur .
→ → → "ind.dpf"
Le fichier .dpf contient les paramètres pour le programme AutoDock. On a choisi l'algorithme génétique Lamarckien comme méthode d'échantillonnage conformationnel.
→ →
Le programme AutoDock est exécuté avec le fichier de paramètres
généré précédemment (la ligne de commande correspondante est
autodock4 -p ind.dpf -l ind.dlg).
→ →
→ → → "ind.dlg"
→ →
→ →
Une barre de contrôle apparaît et permet de parcourir les conformations trouvées par le docking. La conformation 0 est celle du départ.
Changer les options en cliquant sur le bouton "&" de la barre.
Sélectionner "Show Info" et examiner les termes énergétiques affichés.
Parcourir les conformations.
Changer le mode de représentation et de coloration du ligand et du récepteur. On pourra par exemple afficher la surface moléculaire du récepteur pour visualiser la complémentarité de forme avec le ligand.
Se positionner sur la meilleure conformation avec la barre de contrôle.
Un nouvel objet "ind_conf_1" est créé.
Sélectionner cet objet.
Sauvegarder la structure dans un fichier PDB
→ → → "ind_conf_1.pdb" →
La structure expérimentale du complexe a pour code PDB 1HSG.
Documentation en ligne de XPLOR 3.1
Installation de Linux dans une machine virtuelle
Formation à la ligne de commande Linux
Linux Command Line Cheat Sheet