Design computationnel de protéines
- Collaborateurs
- Thomas Simonson et David Mignon
- Laboratoire
- Laboratoire de Biologie Structurale de la Cellule
- Institution
- École Polytechnique
Le design de protéines a pour but la conception de nouvelles protéines ou la modification de protéines existantes pour atteindre une fonction donnée. Les approches computationnelles sont une aide précieuse pour le design de protéines, pour rationaliser les prédictions et guider les tests expérimentaux. Le design computationnel de protéines (CPD) a suscité d'importants efforts méthodologiques et a obtenu des succès spectaculaires comme la création d'une protéine avec un nouveau repliement ou l'ingénierie de sites actifs d'enzymes. La principale difficulté du CPD réside dans le nombre astronomique de séquences et conformations possibles, de l'ordre de (20 × 10)100 pour une protéine de 100 acides aminés. Un autre élément clé pour le succès du CPD est la fonction d'énergie utilisée pour évaluer et sélectionner les séquences et les conformations.
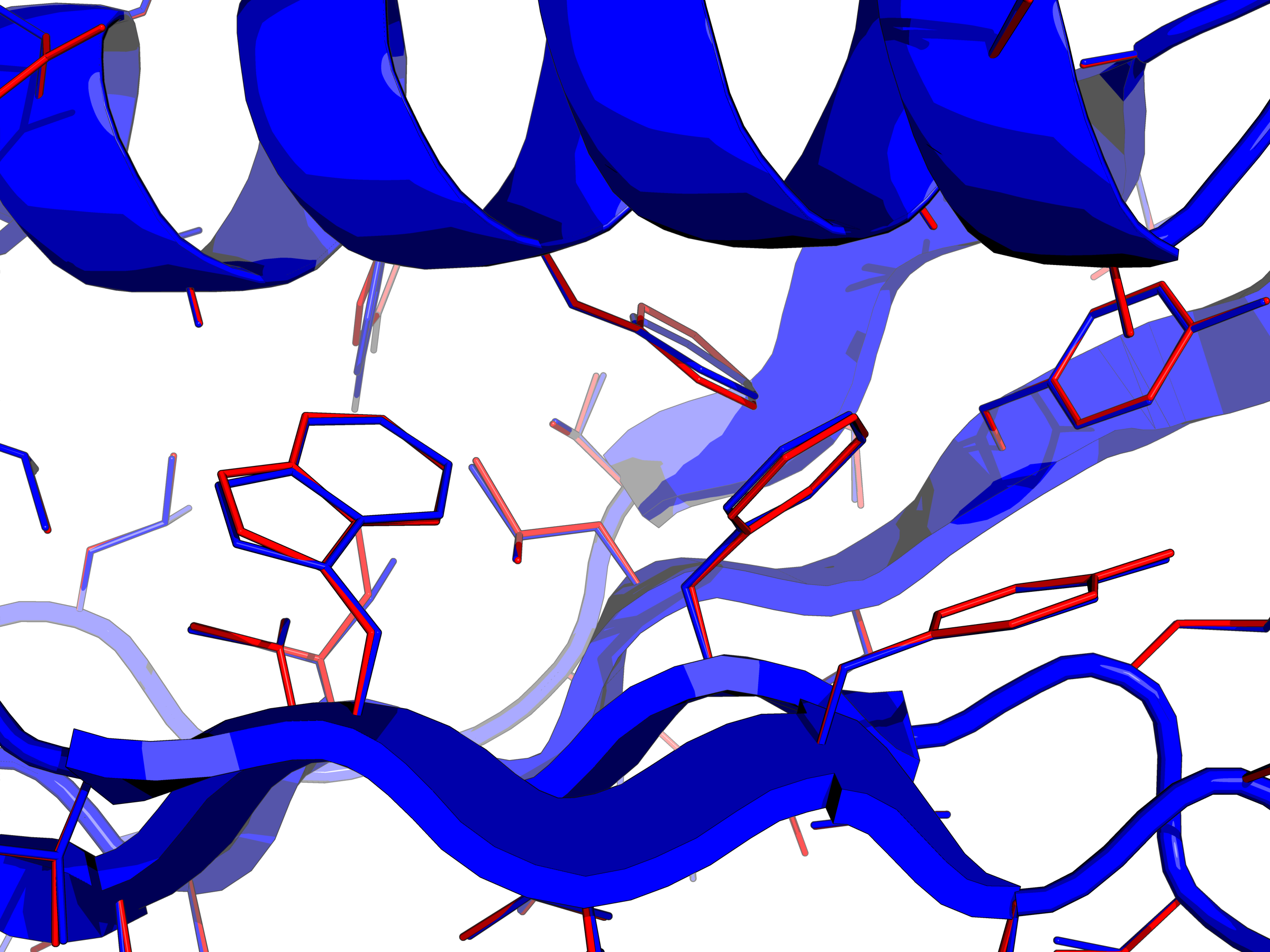
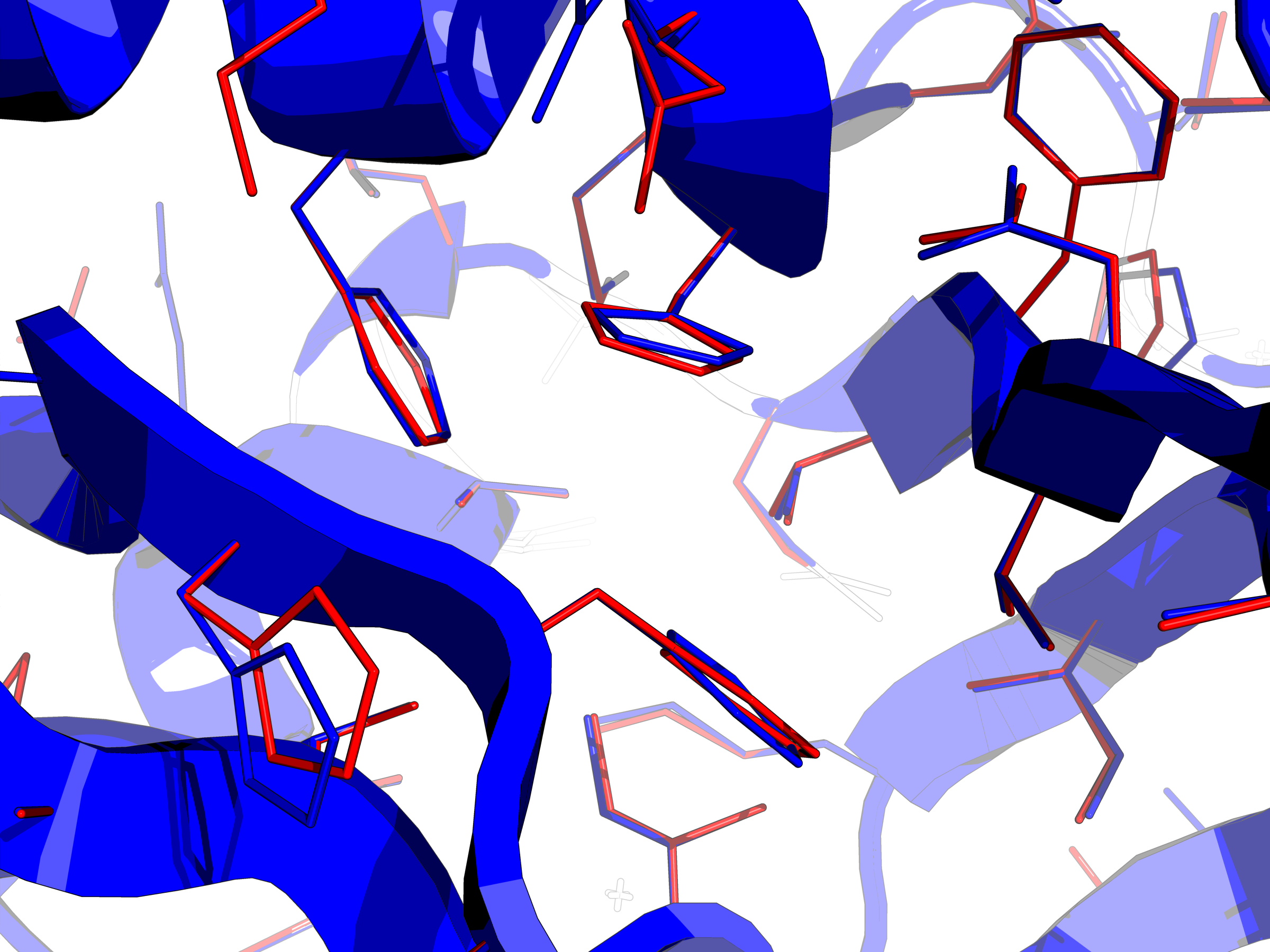
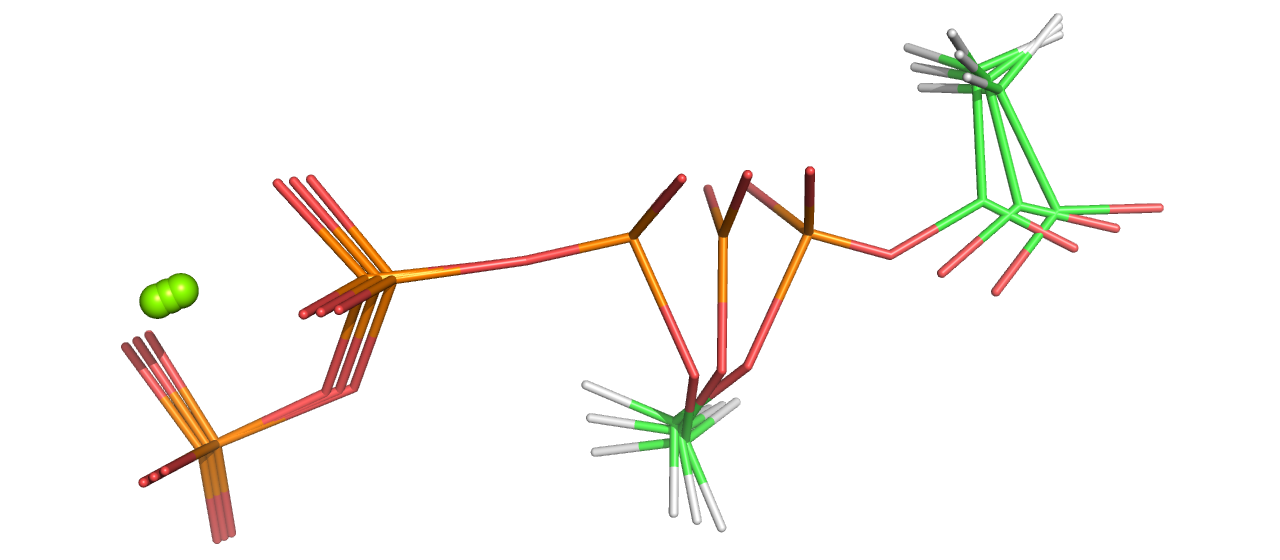
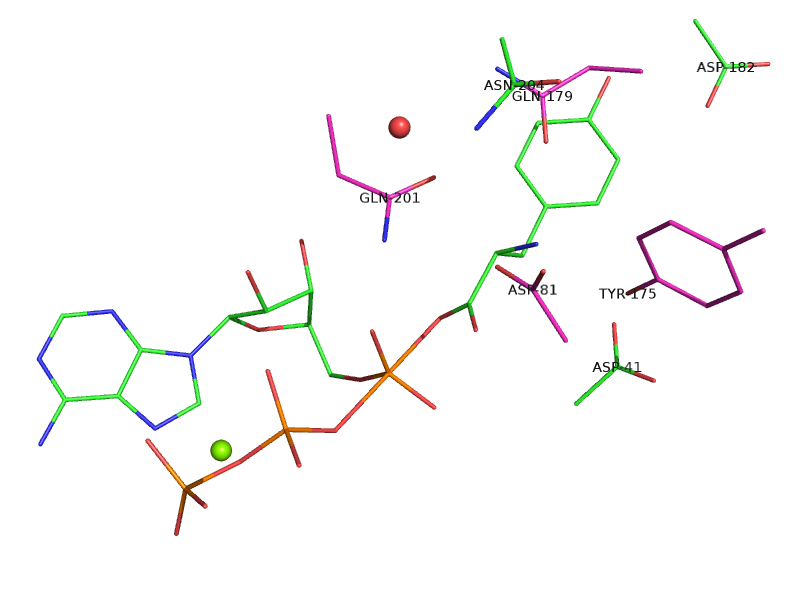
L'équipe de bioinformatique structurale du laboratoire a travaillé pendant une vingtaine d'années sur le CPD et a développé un logiciel appelé Proteus (https://proteus.polytechnique.fr ), en suivant une approche inspirée par la physique. Elle se fonde sur un modèle atomique de la structure de la protéine et sur une fonction d'énergie de mécanique moléculaire. Un aspect important est le traitement du solvant, représenté par un continuum diélectrique avec un terme de Born généralisé, complété par un terme proportionnel à la surface accessible au solvant. Les particularités de l'implémentation sont : 1) le squelette de la protéine est maintenu fixe, 2) l'espace conformationnel des chaînes latérales est réduit à une bibliothèque discrète de rotamères, 3) la fonction d'énergie est décomposée en paires d'interactions. La première étape consiste à calculer une matrice d'interactions entre chaque paire de rotamères. Dans la seconde étape, l'espace des séquences et conformations est exploré avec un algorithme d'optimisation. Les évaluations d'énergie sont rapides dans cette seconde étape grâce au précalcul de la matrice d'énergie. Proteus peut traiter une grande variété de problèmes. Il a été appliqué entre autres à la prédiction de chaînes latérales, à la prédiction de la stabilité de mutants, à la reconnaissance de pli, aux prédictions de pKa, au redesign de séquences entières de protéines, et à l'ingénierie de sites actifs d'enzymes.
Dans ce projet, j'ai contribué plus particulièrement aux développements méthodologiques, notamment aux travaux sur la fonction d'énergie, les modèles de solvatation, leur décomposition en paires, et l'implémentation du calcul de la matrice d'énergie. Par ailleurs, j'ai conduit des applications de ces modèles à différents problèmes de CPD, avec une évaluation de leur performance et de la contribution de leurs composantes.


- Références
-
Transition state-based computational enzyme design.
T. Gaillard, T. Simonson
in S. M. Kahn, F. Pazos (Éd.), Methods in Molecular Biology: Protein Design and Evolution.
Humana, New York, 2026.
doi:10.1007/978-1-0716-4828-5_11 -
Improved Physics-Based Single-Position Protein Sequence Redesign with a Residue-Pairwise Generalized Born Model.
T. Gaillard*
Journal of Physical Chemistry B, 2025, 129, 10699-10710.
doi:10.1021/acs.jpcb.5c03662 -
Physics-Based Computational Protein Design: An Update.
D. Mignon, K. Druart, E. Michael, V. Opuu, S. Polydorides, F. Villa, T. Gaillard, N. Panel, G. Archontis, T. Simonson*
Journal of Physical Chemistry A, 2020, 124, 10637-10648.
doi:10.1021/acs.jpca.0c07605 -
Adaptive Landscape Flattening Allows the Design of Both Enzyme:Substrate Binding and Catalytic Power.
V. Opuu, G. Nigro, T. Gaillard, E. Schmitt, Y. Mechulam, T. Simonson*
PLOS Computational Biology, 2020, 16, e1007600.
doi:10.1371/journal.pcbi.1007600 -
Full protein sequence redesign with an MMGBSA energy function.
T. Gaillard*, T. Simonson*
Journal of Chemical Theory and Computation, 2017, 13, 4932-4943.
doi:10.1021/acs.jctc.7b00202 -
Protein side chain conformation predictions with an MMGBSA energy function.
T. Gaillard*, N. Panel, T. Simonson*
Proteins, 2016, 84, 803-819.
doi:10.1002/prot.25030 -
Pairwise decomposition of an MMGBSA energy function for computational protein design.
T. Gaillard*, T. Simonson*
Journal of Computational Chemistry, 2014, 35, 1371-1387.
doi:10.1002/jcc.23637 -
Computational protein design: the Proteus software and selected applications.
T. Simonson*, T. Gaillard, D. Mignon, M. Schmidt am Busch, A. Lopes, N. Amara, S. Polydorides, A. Sedano, K. Druart, G. Archontis
Journal of Computational Chemistry, 2013, 34, 2472-2484.
doi:10.1002/jcc.23418 -
Computational protein design in the genomic era.
T. Gaillard, M. Schmidt am Busch, A. Lopes, D. Mignon, T. Simonson
Actes des Journées Ouvertes en Biologie, Informatique et Mathématiques, 2013, 151-158.
1-4 juillet 2013, Toulouse, France.
-
The inverse protein folding problem: protein design and structure prediction in the genomic era.
M. Schmidt am Busch, A. Lopes, D. Mignon, T. Gaillard, T. Simonson
in J. Zeng, R. Zhang, H. Treutlein (Éd.), Quantum Simulations of Materials and Biological Systems.
Springer Verlag, New York, 2012.
